


发布时间:2019-07-16 来源: 浏览量:
内容提要
美国黑框警告某些治疗失眠类药物的复杂睡眠行为造成严重伤害的风险
欧盟建议撤回奥拉单抗的上市许可
欧盟建议限制多发性硬化治疗药物阿仑单抗使用
英国警示艾维雷韦/克比司特因治疗失败和母婴传播HIV的风险避免在孕期使用
英国警告免疫功能抑制和60岁以上患者接种黄热病疫苗可能导致死亡的风险
英国警示贝利木单抗严重精神不良事件发生增加的风险
美国黑框警告某些治疗失眠类药物的复杂睡眠行为造成严重伤害的风险
2019年4月30日,美国食品药品管理局(FDA)发布消息称,由于复杂睡眠行为(包括梦游、睡眠驾驶和在不完全清醒的情况下从事其他活动),某些常见的治疗失眠类处方药物发生了罕见但严重的伤害,甚至死亡。与其他用于治疗失眠的处方药相比,这些行为似乎更常见于艾司佐匹克隆、扎来普隆和唑吡坦。因此,FDA要求在这些药品的说明书和患者用药指南中添加黑框警告,同时要求将既往使用艾司佐匹克隆、扎来普隆和唑吡坦曾发生过异常睡眠行为的患者列为禁忌人群。
艾司佐匹克隆、扎来普隆、唑吡坦属于镇静催眠类药物,用于治疗成人睡眠障碍,已批准上市多年。复杂睡眠行为造成的严重伤害和死亡发生在有或没有此类行为史的患者身上,不管是否使用最低推荐剂量或单次用药,无论有没有并用酒精或其他中枢神经系统抑制剂(如镇静剂,阿片类药物,和抗焦虑药物等),使用这些药物均可能发生异常睡眠行为。
针对医务人员信息:
对于服用艾司佐匹克隆、扎来普隆、唑吡坦后出现过复杂睡眠行为的患者应避免开具这些药物;如果患者出现过复杂的睡眠行为,应停止使用这些药物,因为这些药物引起的行为虽然罕见,但已导致严重的伤害或死亡。
针对患者信息:
如患者服药后不完全清醒,或者如果不记得做过的活动,可能出现了复杂的睡眠行为,应停止使用治疗失眠的药物,并立即就医。
在过去的26年里,FDA发现了66例药物引起复杂睡眠行为的报告, 这些病例仅来自FDA不良事件报告系统(FEARS)或医学文献,因此可能有更多尚未发现的病例。66例病例包括意外服药过量、跌倒、烧伤、溺水、暴露在极低的温度下导致肢体功能丧失、一氧化碳中毒、溺水、体温过低、机动车碰撞、以及自伤(如枪伤和明显的自杀企图)。患者通常不记得这些事件。这些治疗失眠药物导致复杂睡眠行为的潜在机制目前尚不明确。
FDA还提醒公众,所有用于治疗失眠的药物都会影响次日早晨驾车及其他需要保持警觉的活动。昏昏欲睡已经被列为所有失眠药物的药物标签上常见的副作用。FDA警告患者服用这些产品后第二天患者仍会感到昏昏欲睡。服用治疗失眠药物的患者在使用后第二天早晨即使感到完全清醒,也会出现精神警觉性下降的现象。
给患者的补充信息
•艾司佐匹克隆、扎来普隆、唑吡坦可引起复杂睡眠行为,包括梦游、睡眠驾驶和在不完全清醒的情况下从事其他活动。这些复杂的睡眠行为是罕见的,但已导致严重的伤害和死亡。
•这些事件只需使用一剂这些药物就可以发生,也可以在较长的治疗时间后发生。
•如果患者出现了复杂睡眠行为,应立即停药,并及时就医。
•按照医嘱用药。为了减少不良事件的发生,不要超剂量、超频次用药。
•如果在服药后无法保证足够的睡眠时间,请不要服用艾司佐匹克隆、扎来普隆、唑吡坦。如果服药后起得太快,可能会感到昏昏欲睡,记忆力、警觉性或协调性出现问题。
使用艾司佐匹克隆、唑吡坦(片、缓释片、舌下片或口腔喷雾剂),应该服药后立即就寝,并卧床7至8个小时。
使用扎来普隆片或者低剂量唑吡坦舌下片,应该在床上服药,并至少卧床4小时。
•服用艾司佐匹克隆、扎来普隆和唑吡坦时,不要同时使用其他任何帮助睡眠的药物,包括一些非处方药。服用这些药物前不要饮酒,因为会增加出现副作用和不良反应的风险。
给医务人员的补充信息
•艾司佐匹克隆、扎来普隆和唑吡坦已经有引起复杂睡眠行为的报告。复杂的睡眠行为是指患者在没有完全清醒的情况下进行活动,可以导致严重的伤害和死亡。
•这些事件只需使用一剂这些药物就可以发生,也可以在较长的治疗时间后发生。
•既往使用艾司佐匹克隆、扎来普隆和唑吡坦出现过复杂睡眠行为的患者,禁止再次处方这些药物。
•告知患者如果出现过复杂睡眠行为,即使没有造成严重的伤害,也要停止使用治疗失眠药物。
•当给患者处方艾司佐匹克隆、扎来普隆或唑吡坦时,应遵循说明书中的剂量建议,从尽可能低的有效剂量开始。
•鼓励患者使用艾司佐匹克隆、扎来普隆或唑吡坦时阅读患者药物指南,并提醒他们不要与其他治疗失眠药物、酒精或中枢神经系统抑制剂联合使用。
(FDA网站)
欧盟建议撤回奥拉单抗的上市许可
2019年4月26日,欧洲药品管理局(EMA)网站发布信息称,建议撤回抗肿瘤药奥拉单抗(Olaratumab,商品名Lartruvo)的上市许可。
EMA完成了对ANNOUNCE研究结果的评估,认为与多柔比星单独用药相比,奥拉单抗与多柔比星联合用药不能延长软组织肉瘤患者的生存时间。因此,EMA建议撤回奥拉单抗的上市许可。
2019年1月,ANNOUNCE研究披露初步结果时,EMA即建议不应对软组织肉瘤新患者启用奥拉单抗治疗。在评估了全部研究结果后,EMA认为不能确证奥拉单抗联合多柔比星的获益。就安全性而言,研究数据没有显示新的安全问题。
奥拉单抗于2016年11月获批上市,用于晚期软组织肉瘤治疗,该病目前缺乏有效治疗药物。在批准上市时,有关奥拉单抗效果的数据有限,支持上市申请的主要临床试验纳入的患者数较少。因此,奥拉单抗是附条件批准上市,企业需提供来自ANNOUNCE研究的补充证据。
给患者的信息
•奥拉单抗曾获批用于治疗软组织肉瘤的罕见肿瘤。其上市许可是附条件批准,企业需开展研究确证该药品的获益。
•但是,上述研究显示,就延长患者生存期而言,与单独使用多柔比星相比,奥拉单抗联合多柔比星没有优势。
•奥拉单抗的上市许可将被撤回,新患者不应启用奥拉单抗治疗。
•如果患者已经开始使用奥拉单抗,经治医生将权衡最恰当的治疗方案。
•没有发现奥拉单抗的新的安全问题。
给医务人员的信息
•奥拉单抗联合多柔比星治疗晚期或转移性软组织肉瘤的III期临床试验ANNOUNCE研究没有确证奥拉单抗的临床获益。
• 上述研究在研究总人群和平滑肌肉瘤亚组中均未达到延长生存期这一主要指标(总研究人群:分层HR 1.05,奥拉单抗联合多柔比星组中位数20.4个月,安慰剂联合多柔比星组中位数19.8个月;平滑肌肉瘤亚组:HR 0.95,奥拉单抗联合多柔比星组中位数21.6个月,安慰剂联合多柔比星组中位数21.9个月)。
•除此以外,对于该研究的其中一项次要指标无进展生存期(PFS),在研究总人群中也未显示出获益(HR:1.23;奥拉单抗联合多柔比星组中位数5.4个月,安慰剂联合多柔比星组中位数6.8个月)
•鉴于以上研究结果,奥拉单抗的上市许可将被撤回,不应对新患者启用奥拉单抗治疗。
•对于已经开始使用奥拉单抗的患者,医生应考虑其它可及的治疗选项。
•上述研究期间未发现新的安全问题。
关于药品的进一步信息
奥拉单抗是2016年11月9日在欧盟批准上市的抗肿瘤药,用于治疗成人晚期软组织肉瘤,一种累及机体软组织例如肌肉、血管以及脂肪组织的恶性肿瘤。
奥拉单抗与多柔比星(另一种抗肿瘤药)联合用药,用于不能接受手术和放射治疗以及既往未接受过多柔比星治疗的晚期软组织肉瘤患者。奥拉单抗与多柔比星联合使用至多8个治疗周期,对于疾病没有恶化的患者,后续可单独使用奥拉单抗。
奥拉单抗属于“附条件批准”(conditional approval)。在批准上市时,有关奥拉单抗疗效的数据有限,支持上市申请的主要临床试验纳入的患者数较少。因此,奥拉单抗获得的上市许可附有条件,即企业需提供来自ANNOUNCE研究的补充证据、以确证该药的获益和安全性。
(欧盟EMA网站)
欧盟建议限制多发性硬化治疗药物阿仑单抗使用
2019年4月12日,欧洲药品管理局(EMA)发布公告称,收到阿仑单抗相关的免疫疾病(由机体防御系统没有正常运转导致)和心脏及血管问题的新的报告后,EMA的药物警戒风险评估委员会(PRAC)启动了一项关于多发性硬化治疗药物阿仑单抗的评估。在评估过程中,作为EMA一项临时措施,阿仑单抗仅可在成人高度活跃的复发缓解型多发性硬化患者中使用,尽管治疗方案中已有至少2种疾病调修药(多发性硬化药物的一种类型),或其他疾病调修药不能使用的情况下。受益于阿仑单抗治疗的患者在咨询医生后可继续治疗。
阿仑单抗用于治疗成人型复发缓解型多发性硬化,该疾病患者中枢神经的环绕神经细胞的保护性髓鞘被炎症损害。复发缓解型意味着患者在较少或无症状期(缓解期)之间有发作(复发)。阿仑单抗用于治疗活动期患者,以静脉输注方式给药。阿仑单抗是单克隆抗体,设计用于识别和攻击免疫系统(人体防御系统)白细胞的CD52蛋白。通过结合CD52,阿仑单抗可导致白细胞死亡和替换,从而减弱免疫系统的破坏性。阿仑单抗于2013年在欧盟上市。
除了上述对阿仑单抗的使用限制,EMA药物警戒风险评估委员会(PRAC)已建议对阿仑单抗的说明书进行更新,以告知患者和医务人员有以下不良反应的相关病例报告:
•免疫相关疾病,包括自身免疫性肝炎(对肝脏损伤)和噬血细胞性淋巴组织细胞增生症(免疫系统过度激活可累及机体不同部位);
•心脏和血管问题发生在接受药物治疗后1-3天,包括肺出血,心脏病发作,中风,颈动脉夹层(头颈部动脉的撕裂);
•严重的中性粒细胞减少(中性粒细胞计数降低,中性粒细胞为抗感染的白细胞的一种类型)
医务人员应对于出现了以上症状的患者需考虑停止其治疗;患者如果出现了上述症状,应尽快就医。
EMA目前将评估所有关于该药品安全性相关的已获得证据,并考虑采取其他的必要措施保护患者,包括是否应对该药品的适应症进行修订。
给患者的信息:
•报告了一些阿仑单抗副作用的新病例,包括一些影响心脏、血管、肺和肝脏的病例。
•如出现以下症状,患者应立刻就医:
—急性(突发)心脏问题(通常在给药后1-3天内发生):例如呼吸困难和胸痛
— 肺出血:例如呼吸困难和咳血
— 中风和给脑供血的血管撕裂:例如面部下垂、突发严重头痛、侧肢体无力、言语困难或颈痛
— 肝脏问题:例如皮肤或眼睛黄染,深色尿,比平常更易出血和淤青
— 噬血细胞性淋巴组织细胞增生症:例如发热、腺体肿胀和皮疹。
• 如果患者出现以上症状,医生将进行检查,可能考虑停止阿仑单抗的治疗,并替换为其他替代治疗方案。
•一项对阿仑单抗的全面深入评估正在进行,进一步的信息将在获得后尽快发布。
•当该项评估正在进行时,阿仑单抗将只被处方给那些其他药物无效或不适合使用的新患者。
•如果有关于治疗的任何问题或担忧,及时与医生沟通。
给医务人员的信息:
•在关于该药物正在进行评估得出结论之前以及在阿仑单抗的产品说明书增加新的安全性警告之前,EMA书面告知医生关于阿仑单抗处方中的临时使用限制。
•新的治疗仅用于那些成人高度活跃的复发缓解型多发性硬化患者,尽管一个全面和充分疗程的治疗方案中已有至少2种疾病调修药治疗,或在那些成人高度活跃的复发缓解型多发性硬化患者中其他疾病调修药治疗禁忌或不适合使用情况下。
•对于正在使用阿仑单抗的患者,在静脉输注阿仑单抗之前和过程中,需要监测生命体征。如果观察到显著的临床指征变化,应考虑中止输注并进行包括ECG在内的额外监测。
•肝功能检测应在治疗之前和过程中开展。如果患者出现了肝损伤症状,非预期的肝酶升高或提示肝功能障碍的症状(例如,不明原因的恶心、呕吐、腹痛,疲劳,厌食,黄疸或棕深色尿),阿仑单抗应仅在谨慎权衡后再次给药。
•患者如果出现病理性的免疫系统激活症状,应立刻评估,并考虑噬血细胞性淋巴组织细胞增生症的诊断。免疫系统异常激活的症状可在治疗后的4年内发生。
•阿仑单抗的评估完成后,将发布更多的信息。
(欧盟EMA网站)
英国警示艾维雷韦/克比司特因治疗失败和母婴传播HIV的风险避免在孕期使用
2019年4月,英国医疗产品健康管理局(MHRA)发布信息称,艾维雷韦/克比司特由于存在治疗失败以及母婴传播HIV的风险应避免在孕期使用。药代动力学数据表明,相比产后,克比司特(Cobicistat)增强的艾维雷韦(Elvitegravir)的暴露量在妊娠II期和III期偏低。艾维雷韦低暴露量可能与治疗失败的风险升高以及母婴传播HIV-1的风险增加有关,因此在怀孕期间不应使用艾维雷韦/ 克比司特。
艾维雷韦是一种整合酶抑制剂,与其他抗逆转录病毒药物联合用于治疗HIV-1。克比司特是一种用于增加艾维雷韦水平的药代动力学增强剂。克比司特可单独作为药物使用,商品名为Tybost。含艾维雷韦/克比司特的药物包括Genvoya(艾维雷韦/克比司特、恩曲他滨/替诺福韦/艾拉酚胺)和Stribild(艾维雷韦/克比司特/恩曲他滨/替诺福韦酯)。
给医务人员的建议:
•药代动力学数据显示,妊娠II期和III期用克比司特增强的艾维雷韦(艾维雷韦/克比司特)暴露量偏低
•虽然迄今为止尚未报告过这种治疗传播的病例,但是低艾维雷韦暴露量可能与治疗失败的风险升高以及母婴传播艾滋病毒感染的风险增加有关
• 怀孕期间不应开始使用艾维雷韦/克比司特治疗
• 将孕妇以及服用艾维雷韦/克比司特的女性转为替代治疗方案
•向黄卡计划报告HIV药物的疑似药品不良反应,包括导致伤害的治疗失败
在2018年7月,MHRA警示孕期不要使用克比司特增强的达芦那韦(darunavir),因为药代动力学数据表明,由于怀孕期间暴露量较低,治疗失败和母婴传播HIV感染的风险增加。同时也评估了含有艾维雷韦/克比司特的治疗风险。来自IMPAACT P1026s(国际母亲儿科青少年艾滋病临床试验P1026研究)的药代动力学数据显示,与配对的产后数据相比,克比司特增强的艾维雷韦血浆浓度24小时后在妊娠II期降低81%,在妊娠III期降低89%。在妊娠II期和III期,克比司特血浆浓度24小时后分别降低60%和76%。
对安全性数据和已发表文献的回顾分析尚未发现在妊娠II期和III期间使用含有艾维雷韦/克比司特治疗妇女的母婴HIV-1传播的病例。然而,由于理论上存在风险,在妊娠期间不应开始使用艾维雷韦/克比司特进行治疗,服用艾维雷韦/克比司特的孕妇也应转为替代治疗方案。
Genvoya和Stribild的产品信息正在更新,建议不要在怀孕期间使用,并已向相关医务人员发送致医生信告知这些风险信息。
(英国MHRA网站)
英国警告免疫功能抑制和60岁以上患者接种黄热病疫苗可能导致死亡的风险
英国药品和健康产品管理局(MHRA)发布信息称,近期收到2例接种黄热病疫苗(商品名:Stamaril)后死亡的不良反应病例。由于存在增加危及生命的不良反应的风险,黄热病疫苗不应用于有胸腺功能障碍病史或免疫功能抑制的患者。同时特别提醒 60岁及以上的患者接种黄热病疫苗前需要仔细进行风险评估,因为有充分的证据表明黄热病疫苗会增加这类人群不良反应的风险。
对医务人员的建议
• 如同其他减毒活疫苗,黄热病疫苗不应用于可能存在免疫功能抑制的患者。
• 黄热病疫苗禁用于有胸腺功能障碍病史(包括重症肌无力和胸腺瘤)的患者。
• 黄热病疫苗禁用于胸腺切除的患者。
• 对于60岁及以上的人群,黄热病疫苗仅用于存在不可避免黄热病感染风险的情况下。
•实施黄热病疫苗接种的专家必须熟知该疫苗的禁忌症和接种前警告。
• 如果怀疑有免疫功能抑制的患者,应推迟接种黄热病疫苗,直至获得专家的建议。
• 应加强接种流程和检查清单管理,以避免因不适当接种而导致的严重的、可能导致死亡的不良反应。接种人员应熟知黄热病疫苗中心业务守则。
• 接种期间任何怀疑的不良反应都应上报。
背景
MHRA 曾在2016年4月的《药物安全最新进展》(Drug Safety Update)中给出建议,并在2017年10月的《药物安全最新进展》中再次提醒,减毒活疫苗不应用于临床上无论是治疗、疾病、还是妊娠导致的免疫功能抑制的患者。因为活的疫苗菌株可以复制,从而导致该类人群广泛的、严重的、有时是致命的感染。
近几个月,MHRA接收到2例接种黄热病疫苗后的死亡病例。其中1例是因胸腺瘤而进行了胸腺切除术(产品说明书中的禁忌症)的患者,另1例是67岁的患者,无已知的风险因素。2例患者接种黄热病疫苗后短期内死亡,怀疑可能是黄热病疫苗导致的嗜内脏型疾病(YEL-AVD)。
YEL-AVD被认为是类似黄热病严重感染的不良反应。全球的报告发生率大约是每1百万接种者中发生1例,有胸腺疾病、免疫功能抑制和60岁及以上患者的风险会增加。另一个严重的接种风险是黄热病疫苗相关的神经性疾病(YEL-AND),该类疾病的发生率和风险因素与YEL-AVD相似。YEL-AND表现为多样的神经系统症状。
黄热病疫苗的警告和禁忌症
有关该疫苗的警告、警示、风险等信息详见产品说明书。黄热病疫苗禁忌用于任何由于治疗或先天性、特发性疾病导致的免疫功能抑制的患者,包括胸腺功能障碍(含重症肌无力和胸腺瘤)病史的患者,胸腺切除术后也属于禁忌症。
由于存在导致严重的、潜在的、致死性不良反应的高风险,对于60岁及以上患者,黄热病疫苗只能用于有不可避免的黄热病感染风险的情况下。
任何处方或实施接种的医务人员必须全面熟知最新的产品说明书。有关该产品的更多信息和指南可以在绿皮书(the Green Book)和国家旅游健康网络和中心(NaTHNaC)的网站中获得。
接种的风险评估
医务人员应当与受种者充分讨论个人风险获益。应预留充足的时间审核用药史、可获得的用药记录,保证患者没有禁忌症,并特别询问是否有任何潜在的胸腺疾病史或胸腺切除史。60岁及以上患者只能在有不可避免的黄热病感染风险的情况下接种。患者信息手册(Patient Information Leaflet)将为潜在的受种者提供有用的基础信息。应使用风险评估检查清单以确保完成每个核对项目以及评估每个患者的免疫功能。NaTHNaC 建议医务人员使用旅行风险评估表来指导旅行健康咨询,如果有特殊用药史则应寻求专业人士的建议。更多信息可在NaTHNaC网站中获得。
MHRA正在对该疫苗开展获益风险评估,并正基于这些病例和最新的科学数据采取风险最小化措施。人类药品委员会(Commission on Human Medicines)正召集专家工作组,以提供相关建议,必要时将更新指南。
(英国MHRA网站)
英国警示贝利木单抗严重精神不良事件发生增加的风险
2019年4月,英国药品和健康产品管理局(MHRA)发布信息称,包括一项随机临床试验在内的临床试验初步结果显示,接受贝利木单抗的系统性红斑狼疮患者与那些除标准治疗外接受安慰剂治疗的患者相比,有增加抑郁、自杀意念/行为、或自我伤害的风险。
贝利木单抗是人源 IgG1λ单克隆抗体,特异性地针对可溶性人B淋巴细胞激活蛋白。贝利木单抗已批准用于已进行了标准治疗的、成人型活跃的自身抗体阳性的且伴随高疾病活动度的系统性红斑狼疮的辅助治疗(例如,阳性的抗dsDNA抗体和低补体情况下)。2018年,在英国贝利木单抗估算暴露量是102个患者年。
在利木单抗获批的临床研究中,与对照组相比,观察到精神不良事件发生率的失衡。评估发现贝利木单抗的获益大于风险。然而,作为附条件的上市许可,贝利木单抗的上市许可持有人被监管机构要求开展一项随机、安慰剂对照的临床试验(BEL115467),以评估本品的全因死亡率和预先指定的特别关注的不良事件,包括选定的严重精神不良事件。目前,该研究正在全球范围内开展。
该项研究的1年期数据初步显示,与标准治疗加安慰剂治疗的患者相比,标准治疗加贝利木单抗治疗患者报告更多的严重抑郁、自杀意念或行为及自我伤害(见表格)的不良事件(见下表)。
表:报告严重抑郁或自杀倾向的患者情况
(治疗人群中,编号BEL115467研究)
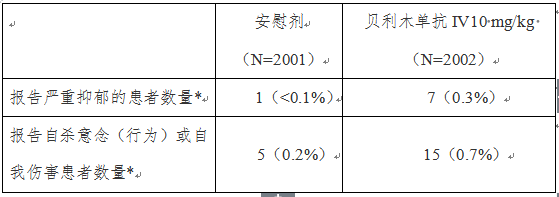
*根据研究者报告
基于以上研究结果,在贝利木单抗治疗之前,建议考虑患者病史和目前精神状态,处方医生应仔细评估患者抑郁和自杀风险;如果患者出现新的或精神症状加重,建议患者尽快就医。
给医务人员建议:
• 包括一项随机临床试验在内的临床试验初步结果显示,接受贝利木单抗治疗的患者有增加严重精神不良事件(抑郁、自杀意念/行为[包括由自杀导致的死亡]、或自我伤害)风险。
• 在贝利木单抗治疗之前,考虑患者病史和目前精神状态,应仔细评估抑郁和自杀风险。
• 在治疗过程中,监测所有患者上述风险相关的新的或加重的症状。
•如果患者出现了新的精神症状或精神症状加重,应评估继续治疗的获益和风险。
给患者(和看护者,如适用)建议:
• 正在使用贝利木单抗的患者,可能经历情绪或行为变化,如果出现了新的或加重的抑郁、自杀意念、或关于伤害自己的想法时应尽快就医。
• 患者可让家庭成员或朋友知悉他们正在使用贝利木单抗,这样家庭成员或朋友可以注意患者情绪上的任何变化。
(英国MHRA网站)
 友情链接
友情链接
 返回列表页
返回列表页
 关闭窗口
关闭窗口
 打印本页
打印本页